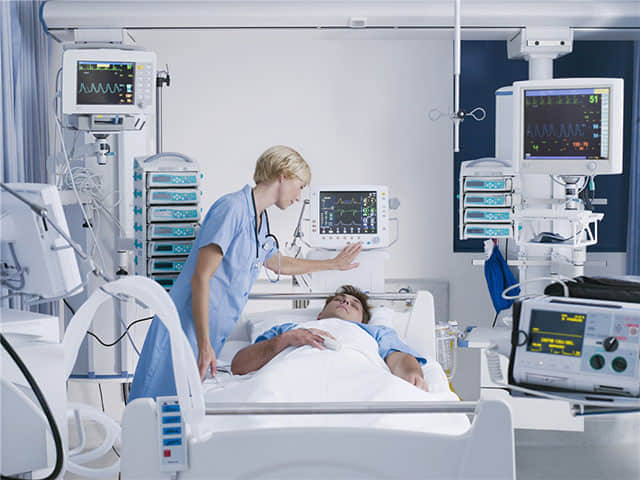
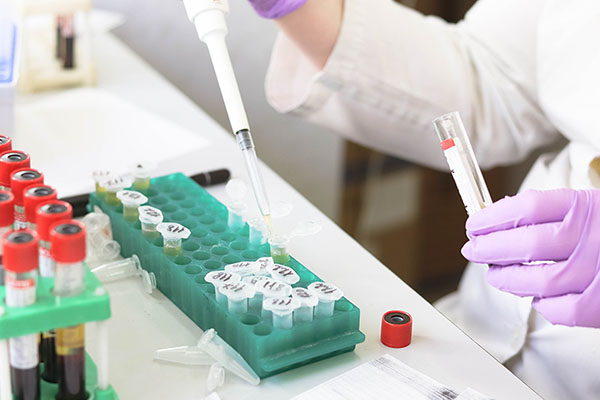
医疗器械的FDA认证是指通过美国食品药品监督管理局(FDA)的审批程序,获得在美国市场上销售和使用的合法授权。这是医疗器械制造商必须遵循的重要法规,以确保其产品在安全和有效性方面符合美国的监管标准。医疗器械
自2021年5月起,欧盟医疗器械新法规2017/745(MDR法规)正式实施,意味着医疗器械应满足MDR法规并获得CE标志批准后,方可在欧洲经济区的31个国家/地区销售设备。
目前英国UKCA认证仍处于过渡期,在2023年7月1日前,中国厂家可以通过原有的CE证书,联系IVDEAR快速完成英国注册。2023年7月1日后,英国将全面启动UKCA认证,原有的CE证书将得不到英国认可,必须通过英国的UKCA公告
CTDA注册具备一定的申请门槛,部分企业可能达不到申请条件的要求。企业必须持有CE或UKCA或MHRA。如不符合此要求,申请将被自动拒绝。企业申请CTDA,对于临床也有一定的要求。临床性能验证阳性样本至少100例,建议
FDA要求所有在美国上市的医疗器械产品都需要进行FDA官网注册,根据产品的不同类别要求如下:-设施注册/公司注册(Establishment,Owner/OperatorRegistration)-产品注册/产品列名(MDL:MedicalDeviceListing)
众所周知,自测CE认证最大的困难点就在于如何实施与完成临床试验研究及可用性研究。 医正通能高效制定出一版有效、靠谱的可行性方案,确保完成后的临床报告与可用性研究报告能得到公告机构的百分百认可。其次,公告
医疗器械认证相关疑问,欢迎您垂询:+8613802989603中国如何定义医疗器械?《医疗器械监督管理条例》第八章附则中医疗器械定义是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类
联系地址:广州市白云区机场路1438-2号正阳大厦8楼A233房
Copyright © 广州医正通医疗管理有限公司
网站地图:sitemap 粤ICP备2024296181号